
SOLVENTES
Definición
Un disolvente o solvente es una sustancia que
permite la dispersión de otra sustancia en esta a nivel molecular o iónico. Es
el medio dispersantes de la disolución.
Normalmente, el disolvente establece el estado físico de la disolución, por lo que se dice
que el disolvente es el componente de una disolución que está en el mismo
estado físico que la misma. Usualmente, también es el componente que se
encuentra en mayor proporción.
Los disolventes forman parte de múltiples aplicaciones: adhesivos,
componentes en las pinturas, productos farmaceúticos, para la elaboración de
materiales sintéticos, etc.
El
papel del solvente no es pequeño. El estudio de reacciones heterolíticas en
ausencia de un solvente en fase gaseosa ha dado un patrón que muestra lo
considerables que pueden ser los efectos del solvente.
La
presencia de un solvente puede acelerar o incluso retardar una
reacción en un factor de 1020; un cambio de un solvente a otro puede
modificar la velocidad de la reacción en casi un millón de veces. Los efectos
de este pueden ser más poderosos que los otros factores; mucho más que los
efectos polares o estéricos, e incluso algo más poderosos que los
efectos de sinforia.
La
selección de un solvente en particular puede ser el factor más importante para
determinar la rapidez de una reacción e incluso si se realiza o no;
puede determinar cuál de los distintos caminos alternativos seguirá
realmente una reacción.
Evidentemente,
un disolvente no es simplemente un lugar, es
una especie de gimnasio donde las moléculas del soluto pueden brincar y
chocar ocasionalmente. El disolvente está íntimamente implicado en toda
reacción que se realiza en él, por lo que para nosotros es
importante comprender cuánto está implicado y de qué modo.
Las moléculas e
iones del soluto no existen en solución como partículas desnudas;
están solvatadas .Hay muchas moléculas de solvente unidas por enlaces
a cada partícula disuelta y es la formación de enlaces la que
proporciona la energía necesaria para que se rompan los enlaces que mantienen
unidas las partículas del soluto. Los solutos apolares
disuelven las sustancias apolares por interacciones entre dipolos inducidos.
El agua es habitualmente denominada el disolvente universal por la gran cantidad de sustancias
sobre las que puede actuar como disolvente.
Actualmente, el empleo de disolventes es masivo en todo tipo de
industrias, lo que plantea problemas de contaminación del medio ambiente (retardan
la división celular y el crecimiento del plancton, producen la muerte de gran
cantidad de organismos) y repercusiones sobre la salud humana.
Los peligros que presenta el uso y manipulación de estos productos
para la salud, han motivado la adopción de una serie de normas de seguridad
(por ejemplo, los límites permisibles de concentración de productos químicos en
el trabajo) para evitar enfermedades e intoxicaciones a los operarios que los
utilizan.
Clasificación
A. Disolventes polares:
Los disolventes polares son aquellos cuyas moléculas constituyentes tienen
regiones densas electrónicamente (bueno, con grandes momentos dipolares y
mayores constantes dieléctricas), y por lo tanto son más fáciles de solvato
(crear una capa sobre el soluto) cualesquiera características de la sustancia
también polar. Algunos de los disolventes polares son mejor conocidos: agua
(disolvente universal), etanol y ácido acético. Sin embargo, el etanol (alcohol
etílico) a menudo se considera un trastorno bipolar se disuelven fácilmente las
sustancias orgánicas no polares.
Los disolventes
polares se pueden subdividir en:
1. Disolventes polares próticos:
contienen un enlace del O-H o del N-H.
2. Disolventes polares
apróticos: son disolventes polares que no tiene
enlaces O-H o N-H. Este tipo de disolvente que no dan ni aceptan protones.
Características
de los disolventes próticos y apróticos
Los disolventes
próticos son moléculas en general muy polares que contienen protones (H+)
ácidos y por lo tanto pueden formar enlaces de hidrógeno con
los solutos. Los disolventes apróticos son aquellos que no contienen hidrógenos ácidos,
por lo cual no pueden formar puentes de hidrógeno.
En la siguiente
tabla, se establece una clasificación general de los disolventes considerando
su carácter polar o apolar y la capacidad para ceder protones y formar enlaces
de hidrógeno. Se muestran las propiedades que presentan los compuestos próticos
y apróticos.
Tipo De Disolvente
|
Características
|
Próticos
|
Poseen un
grupo funcional capaz de ceder protones (OH, NH, SH).
Capacidad
de formar puentes de hidrógeno.
Polares.
Ejemplos: agua, ácidos carboxílicos, alcoholes, aminas.
|
Apróticos polares
|
Carecen de
grupos funcionales capaces de ceder protones.
Constante
dieléctrica alta.
Ejemplos: DMSO, DMF, HMPA, nitrilos, cetonas, nitrocompuestos.
|
Apróticos apolares
|
Carecen de
grupos funcionales capaces de ceder protones.
Constante
dieléctrica baja.
Ejemplos: hidrocarburos (alifáticos, aromáticos, halogenados), éteres, ésteres, halogenuros de alquilo.
|
B. Disolventes apolares:
En general son sustancias de tipo orgánico y
en cuyas moléculas la distribución de la nube electrónica es simétrica; por lo
tanto, estas sustancias carecen de polo positivo y negativo en sus moléculas.
No pueden considerarse dipolos permanentes. Esto no implica que algunos de sus
enlaces sean polares. Todo dependerá de la geometría de sus moléculas. Si
los momentos dipolares individuales
de sus enlaces están compensados, la molécula será, en conjunto, apolar.
Algunos
disolventes de este tipo son:
El dietiléter, benceno, tolueno, xileno, cetonas, hexano, ciclohexano, tetracloruro de carbono es
el que disuelve o va a disolver, o también el diablo rojo, etc. El cloroformo
por su parte posee un momento dipolar considerable debido a que las poseer tres
cloros en su molécula de carácter electronegativo, hace que el carbono adquiera
una carga parcial positiva y el Hidrógeno una carga parcial negativa, lo que le
da cierta polaridad. Un caso especial lo constituyen los liquidos fluorosos,
que se comportan como disolventes más apolares que los disolventes orgánicos
convencionales.
Los
disolventes más utilizados actualmente, son
los disolventes orgánicos, que son compuestos volátiles que
se utilizan solos o en combinación con otros agentes, sin sufrir ningún cambio
químico, para disolver materias primas, productos o materiales residuales, o se
utilice como agente de limpieza para disolver la suciedad, o como disolvente, o
como medio de dispersión, o como modificador de la viscosidad, o como agente
tenso-activo. El uso de estos disolventes, libera a la atmósfera compuestos
orgánicos volátiles (COVs), que tienen algunos problemas importantes para el
entorno. Algunos COVs causan la degradación de la capa de ozono como es el caso de
1,1,1-tricloroetano, tetracloruro de carbono, CFCs, HCFCs. Entre los solventes
orgánicos más destacados podemos encontrar metanol, etanol, acetona,
cloroformo, tolueno o el xileno, entre otros.
Clases
de disolventes alternativos
1.
Disolventes reactivos:
Son disolventes con baja volatilidad relativa, y durante la formación de
película tienen la capacidad de reaccionar con otros componentes de la pintura,
por ello no se evaporan al medio ambiente (no constituyen COVs), tal como los
coalescentes reactivos (en pinturas base agua) y diluyentes reactivos (en
pinturas epóxicas).
2.
Disolventes benignos:
Se designa como “disolventes benignos” aquellos disolventes ambientalmente
amigables y cuyo grupo incluye disolventes libres de compuestos clorados, con
baja toxicidad y baja RIM (reactividad incremental máxima), comparados con los
disolventes convencionales.
3.
Disolventes Neotéricos:
El término neotérico designa nuevo, moderno, reciente y en este caso hace
referencia a una nueva generación de disolventes, desarrollados de acuerdo a la
filosofía de la “Química verde”. Son una serie de disolventes que presentan una
menor toxicidad, son más seguros y menos contaminantes que los disolventes
convencionales.. Entre ellos se incluyen
tanto nuevos fluidos con propiedades ajustables, como compuestos poco usados
como disolventes en la actualidad. Pero que están siendo investigados por sus
usos potenciales como disolventes, ya que permitirían una mayor sostenibilidad
en futuras aplicaciones. Éste es el caso del dióxido de carbono supercrítico
(scCO2), y el líquido iónico a temperatura ambiente.
Clasificación de los disolventes neotéricos
- Líquido
fluoroso: Son térmicamente estables, por lo que se pueden usar a altas
temperaturas sin peligro. No son inflamables ni tóxicos, por lo que se
evita los riesgos de incendios o explosiones. Se pueden mezclar con muchos
disolventes orgánicos y con el agua, lo que facilita el uso de sistemas
bifásicos.
- Líquido
iónico: Son compuestos que
presentan características de sales con su punto de temperatura de fusión
por debajo de los 100ºC. Compuestos por un catión orgánico siendo uno de los mas comunes el tetraalquilamonio, y un aniónpoliatómico como puede ser el hexafluorofosfato. Los líquidos iónicos pueden ser
constituidos por un gran número de aniones y cationes con lo que sus
propiedades varían de unos a otros. En cuanto a la utilización de un Liquido iónico como disolvente alternativo, reseñar que presenta una escasa
volatilidad, debido a su presión de vapor prácticamente nula. También
presenta una excelente estabilidad química y térmica, pudiéndose emplear a
elevadas temperaturas.Y una gran solvatación con otras muchas sustancias.
En contra partida decir que su coste de obtención es elevado.
- Fluido
supercrítico: Por su importancia como disolvente alternativo cabe destacar
el fluido supercrítico, que es aquel que se encuentra por encima de su
presión y de su temperatura crítica. En este estado, la
línea de separación de fases líquido-gas se interrumpe. Esto implica la
formación de una sola fase, en la que el fluido tiene propiedades
intermedias entre las de un líquido y las de un gas, por lo que mientras
se mantiene una gran difusividad propia de los gases, se consigue una alta
densidad cercana a la de los líquidos. Considerados como inertes y no
tóxicos, siendo su coste barato y pudiendo variar sus propiedades con
cambios de presión. La propiedad más característica de los fluidos
supercríticos es el amplio rango de altas densidades que pueden adoptar
dependiendo de las condiciones de presión y/o de temperatura, a diferencia
de los líquidos que son prácticamente incompresibles, y de los gases que
poseen densidades siempre muy bajas.
La
clasificación química de los disolventes:
A. Disolventes
hidrocarbonados o hidrocarburos: son compuestos
naturales orgánicos, Formados a altas temperaturas y presión durante varias
eras geológicas. Su núcleo básico elemental es el carbono y el hidrógeno.
Se
encuentran en la naturaleza:
-
Formando parte de los
petróleos.
-
Asfaltos naturales.
B. Disolventes no hidrocarbonados: son aquéllos cuyo elemento básico
no es el hidruro de carbono (CH4). El único disolvente industrial que no es
hidrocarbonado es el disulfuro de carbono (CS2).
También cabe diferenciar dentro de los
disolventes:
·
Disolventes puros: con
un solo compuesto químico puro.
·
Disolventes simples:
con un solo compuesto generalmente no puro por contener impurezas de otros.
·
Disolventes compuestos
o mezclas: contienen mezclados varios disolventes de forma intencionada para
las diferentes aplicaciones.
I.- Migración de
los solventes
A. Gravedad, hidrostática,
hidrodinámica: Los líquidos están constituidos
por moléculas distantes las unas de las otras en alrededor de 10 °A. Esta distancia corresponde a interacciones no
despreciables que les permiten rodar las unas sobre las otras permaneciendo prisioneras del
conjunto.
Es por esta razón que los líquidos
adoptan la forma de los recipientes que los contiene.
Esta
distancia de 10 Å es
suficientemente pequeña para que sea difícil comprimir aún más estas moléculas
entre sí. Los líquidos son prácticamente incomprimibles. Esto explica el
fenómeno de Pascal:
“Toda
presión ejercida en la superficie de un fluido se transmite integralmente en
todos los sentidos”.
De
esta forma, la presión ejercida en la superficie de una pintura por un hisopo
será transmitida en todos los sentidos por el solvente, lo que puede
eventualmente provocar levantamientos a distancia.
B. Viscosidad:
Las moléculas de un fluido pueden escurrir las unas sobre las otras, pero sólo en
los fluidos llamados perfectos, lo hacen
sin roce. De hecho, los líquidos siempre presentan un grado de roce: Por esto
son llamados viscosos.
Se
puede visualizar un fluido que escurre, como una serie de paralelas que se
resbalan, las unas sobre las otras con
un roce y arrastre mutuo.
La
rapidez de escurrimiento es máxima en el centro del conducto. En una
canalización de sección delgada, la viscosidad provoca una disminución
progresiva de la presión del líquido. Se denomina como pérdida de carga.
Los
solventes más viscosos serán los más frenados, lo que dificultará su
penetración.
Se
han medido las viscosidades de algunos solventes comunes. A partir de estas mediciones, los solventes
pueden ser clasificados en solventes viscosos (más de 10 cp), intermedios (de 2
a 10 cp) y fluidos (menos de 2 cp a 20°C).
Cuando
se mezclan varios solventes, es muy difícil prever cual será la viscosidad
final. Según las proporciones de los constituyentes de una mezcla, puede
existir una variación regular de la viscosidad pero ocurre que una mezcla
presenta un máximo o un mínimo de viscosidad para una composición dada.
C. Escurrimientos
laminares y turbulentos: Un fluido que escurre
sin obstáculo en un conducto muy liso y cilíndrico presenta normalmente un régimen
llamado laminar. La velocidad de escurrimiento crece desde las paredes hasta el
eje del recorrido, donde alcanza su máximo valor. Este caso es muy raro en los
materiales componentes de obras de arte.
El
más mínimo obstáculo, ya sean paredes rugosas, conductos sinuosos y cambios de
dirección, perturban la velocidad del escurrimiento que se vuelve irregular.
Este tipo de escurrimiento es denominado Turbulento.
Puede
existir también una combinación de ambos regímenes, llamado mixto.
La
turbulencia provoca una pérdida de carga más importante que en un régimen
laminar, pero favorece la mezcla de diferentes compuestos del líquido, lo que
conlleva a una disolución de las paredes del cuerpo poroso.
D.
Interacción
en superficies:
1. Tensión
superficial: Las superficies de los líquidos presentan propiedades muy
particulares. Se comportan similares a
una membrana elástica estirada.
Al
interior de la masa líquida las interacciones están dirigidas en todos los
sentidos y se compensan en la superficie en cambio, hay interacciones dirigidas
hacia el exterior que no son compensadas, estas fuerzas tienden a hacer la
superficie lo más densa posible y será necesario vencer a estas fuerzas para
romper la superficie.
Se
denomina Tensión superficial a la fuerza que mantiene en contacto las moléculas
de la superficie de un líquido, sobre una extensión de 1 cm.
El
valor de la tensión superficial varía notoriamente según la naturaleza del
líquido. Es el agua la que presenta el valor más elevado. Esta característica
se relaciona con las fuertes interacciones que existen entre las moléculas de
agua.
2. Mojado:
Mientras más alta es la tensión superficial de un fluido, mayor será la
tendencia a aglutinarse en gota y más difícilmente mojará un sólido θ Las
fuerzas de interacción de las moléculas del líquido compiten con las fuerzas de
interacción de las moléculas del sólido. Cuando estas últimas ganan, el mojado
es bueno, y el ángulo de mojado es pequeño.
3. Estructura
de la interfase: La superficie de un líquido presenta, en razón de la tensión
superficial, otra estructura que las capas profundas. Se sabe que ciertas
moléculas se comportan como pequeños imanes:
El centro de
acción de las cargas positivas y el de las cargas negativas no coinciden, son
las moléculas llamadas POLARES. Las superficies de los líquidos polares están
formadas por moléculas orientadas de forma muy precisa: En los alcoholes por ejemplo,
los grupos hidróxilos OH, se dirigen hacia el interior del líquido.
En el caso de las
moléculas no polares, como el tetracloruro de carbono, no hay orientación.
El
benceno que no es totalmente simétrico podría adoptar una posición privilegiada
en plano.
E.
Capilaridad:
El fenómeno de capilaridad fue
constatado por primera vez por Leonardo Da Vinci: Se dio cuenta que el agua se
elevaba por conductos de pequeño diámetro (inferiores a 1 mm).
Este
fenómeno es descrito por la ley de Jurin: “La altura hasta donde se eleva un
líquido en un capilar varía en sentido inverso al radio del tubo, en el lugar
donde se detiene el líquido y en proporción directa con la tensión
superficial”.
F. Adsorción :
La adsorción es un fenómeno según el cual substancias gaseosas, líquidas o
sólidas quedan fijadas a la superficie de ciertos sólidos o líquidos sin que
haya formación de compuestos químicos definidos
G. Difusión:
desplazamiento de átomos o moléculas por dos líquidos se van a mezclar, aunque
no se agiten; más o menos rápidamente. El mismo fenómeno ocurre entre sólidos
de masa volumétrica diferente, pero mucho más lentamente.
La
velocidad de difusión depende del tamaño de las partículas migratorias. En una
solución, el solvente y el soluto están divididos hasta
la escala molecular 10 - 3 (m).
Cuando el tamaño de las partículas alcanza de 10 -3 a 0.2 (m), tenemos una
dispersión coloidal. La dispersión de partículas más grandes es mucho más lenta
que la de las moléculas o iones en solución verdadera.
Cuando
una difusión se realiza a través de los agujeros de una membrana, sólo las
partículas suficientemente pequeñas logran pasar. Las más grandes quedan
bloqueadas: Se produce una diálisis.
Cuando
se limpia un cuadro que tiene capas de pintura al aceite de cierta edad, la
difusión y la diálisis provocarán la eliminación por parte del solvente de
pequeñas moléculas como glicéridos y
ácidos grasos de pequeño peso molecular, formados en el transcurso del envejecimiento
de los materiales y que son los que permiten la elasticidad de la película. La acción
de los solventes hará la pintura más quebradiza.
Si soluciones de concentraciones diferentes
son separadas por una membrana en dos compartimentos, se constata que un solvente va a migrar de un
compartimento al otro de forma de igualar las
concentraciones: Es el fenómeno llamado Osmosis.
Este
fenómeno puede provocar a su vez desplazamiento de solventes. El solvente sube
a través de la membrana entrando en un tubo contenedor de solución. La altura de la columna en el tubo da la medida
de la presión osmótica. En la pintura,
pareciera que el fenómeno de osmosis sería el principal responsable de la
formación de ampollas por transferencia de agua
II.- Evaporación
La
velocidad de evaporación de un solvente es una de las dudas más comunes y más
difíciles de resolver por los restauradores y químicos. Esta velocidad de
evaporación depende de un gran número de factores a veces contradictorios entre
sí.
A.
Presión
o tensión de vapor de saturación: Un líquido
ubicado en un recipiente abierto o extendido en una superficie, se evapora
progresivamente hasta que todas las moléculas en estado líquido hallan pasado
al estado vapor. Si se cierra herméticamente el recipiente, se establece un
equilibrio entre el líquido y su vapor.
Después de cierto tiempo, el vapor alcanza una
presión característica para cada líquido a una temperatura determinada: Es la
presión de vapor de saturación, que corresponde al número de moléculas que pasa
del estado vapor al estado líquido e inversamente se equilibran para mantener
esta presión constante a temperatura determinada.
Mientras
más elevada es la presión de vapor de saturación, más moléculas en estado de
vapor existirán en un recipiente cerrado. En un recipiente abierto, el líquido
se evaporará por lo tanto mucho más rápido.
B.
Ebullición:
La presión (o tensión) de vapor aumenta
con la temperatura. Así la temperatura
de ebullición de un líquido corresponde al valor de la tensión de vapor, que es
igual a la presión exterior sobre este líquido, cual sea el valor de esta
presión.
La
ebullición es en efecto, una forma particular de evaporación que se produce en
el interior de un líquido.
En
una mezcla de solventes, la temperatura a la cual la presión de vapor alcance
el valor de la presión exterior, dependerá de la composición de la mezcla.
Normalmente, es el compuesto más volátil el que se evapora más rápido y el
líquido se enriquece con el compuesto menos volátil. La T° de ebullición
entonces aumenta progresivamente.
En
una mezcla, puede ocurrir que debido a fuertes interacciones intermoleculares,
se forme lo que se denomina azeótropos. Es una mezcla que hierve a temperatura
constante y que por lo tanto es imposible de separar por destilación.
Corresponde a proporciones muy determinadas de cada solvente.
La
temperatura de ebullición es una de las primeras características para
clasificar los solventes.
C.
Calor
latente de evaporación
En
un espacio mantenido a una T° constante. Colocar ahí un recipiente que contiene un líquido sellado por un pistón (bien
hermético). Si se levanta lentamente el pistón, el líquido pasará
progresivamente al estado de vapor y en un momento determinado no quedará
ninguna gota de líquido.
A todo lo largo
de esta evaporación, el termostato ha tenido que aportar el calor necesario
para mantener la T° constante, ya que la evaporación conlleva un enfriamiento.
La
cantidad total de calor aportado para que un gramo de líquido sea convertido
totalmente al estado de vapor constante es llamado calor latente de evaporación
de ese líquido. Varía según la presión atmosférica.
Se
ha constatado que los valores más altos de T° de vaporización son los del agua
y alcoholes. Esto se explica por las muy fuertes interacciones intermoleculares
que existen en estado líquido para estos compuestos, y particularmente a los
enlaces de hidrógeno.
D.
La
conductividad térmica
El
calor se transmite de un cuerpo caliente a un cuerpo frío o de una zona más
caliente a una zona más fría de un mismo cuerpo. Este proceso ocurre más o
menos rápido según la capacidad de ese o esos cuerpos de ser más o menos
conductores del calor. Ciertas substancias como los metales son muy conductores,
otros como el vidrio y la madera lo son menos y pueden servir de aislante
térmico.
Un
solvente que para evaporarse necesita un aporte de calor, lo tomara mucho más
fácilmente del medio ambiente si es conductor del calor.
Es
necesario tomar en cuenta que si la evaporación ocurre muy bruscamente, esto
provocará un fuerte y brusco enfriamiento que puede frenar la continuación del
proceso.
Todos
los solventes poseen una conductibilidad térmica superior a la del aire. Al
llenar las fisuras y los poros en una limpieza, estos mejoran espontáneamente
la conductibilidad térmica del cuerpo poroso.
Relaciones
Termodinámicas
Se
conoce que la termodinámica estudia los intercambios de energía, que ocurren
durante el curso de un fenómeno entre un sistema y el medio externo (por
ejemplo la disolución de un barniz o la vaporización de un solvente).
Esta se apoya en
dos principios:
1. Primer principio:
Conservación de la energía: la
cantidad total de la energía es constante, cuales seas las transformaciones de
esta energía al interior del sistema. La energía puede en efecto presentarse de
varias formas: mecánica, química, térmica, eléctrica, etcétera. El principio
puede enunciarse de la siguiente manera: en un sistema aislado que sufre transformaciones, la energía se
conserva pero se degrada.
2. Segundo principio:
entropía: la mayoría de las
transformaciones se hacen en sentido privilegiado, este sentido corresponde a un
aumento del desorden se hace constatado. Que cuando un sistema puede
evolucionar espontáneamente de diferentes formas, es la que provoca el mayor
aumento del desorden la que se producirá.
La
variación de la entropía traduce la variación del desorden.
Cuando
un solvente disuelve un soluto, las moléculas del solvente son separadas las
unas de las otras y es así también para las moléculas del soluto. Se debe
vencer la energía de cohesión que existía centre estas moléculas y
reemplazarlas por nuevas interacciones mixtas solvente soluto.
Esta
operación de mezcla necesita cierta energía que se denomina calor de mezcla
(entropía).
Existe otro concepto de termodinámica: la
energía del calor de la mezcla y de la entropía, cuyo comportamiento indica si
existirá o no disolución.
Expresando la siguiente ecuación:
∆F = ∆H –T ∆S
En 1916, Joel Hildebrand después de hacer diversos experimentos sobre
la solubilidad, formuló el principio de que:
La variación de la energía libre de disolución de un
polímero en un solvente debe ser negativa para que haya disolución, es decir la
conlleva una disminución de la energía libre se puede considerar que los
polímeros presentan en las pinturas (barniz) están en estado amorfo (no
cristalino) y que su disolución puede ser considerada como una simple mezcla.
En un solvente líquido el estado del desorden es
relativamente grande ya que las moléculas pueden escurrir las unas sobre las
otras, aunque permanezcan prisioneras de la masa liquida pueden también dejar
la fase liquida para pasar a la fase de vapor.
Las grandes moléculas de polímeros por el contrario
son mucho menos móviles a causa de su tamaño y forma.
Cuando se mezclan las pequeñas moléculas de solvente
con estas grandes moléculas, estas últimas aceptan un aumento de entropía. En
efecto, se tornan más libre para no moverse según su viscosidad y pueden
ubicarse de otra forma (entropía de configuración) ya que la nueva libertad de
rotación alrededor de los enlaces C-C permite a la molécula adoptar diferentes
formas.
De esto resulta que la disolución es acompañada de
una gran variación de la entropía y el factor –T ∆S (variación de la entropía y
la temperatura absoluta) tiende a hace negativa la variación de la energía
libre.
Todavía
hay que determinar H que mide el calor de la mezcla.
Hildebrand supone que la energía necesaria para que
las moléculas de soluto puedan ser separadas entre sí por las moléculas de
solvente puede ser medida por el calor
de vaporización, ya que también hay separación de moléculas en el transcurso de
la vaporización.
Si la energía necesaria para separar las moléculas A
las unas de las otras es muy diferente a la necesaria para las moléculas B,
cada tipo de molécula preferirá sus similares y no se mezclara con las otras.
Por el contrario, si las energías son del mismo orden de magnitud, las A
toleraran a las B en su proximidad y viceversa. En este caso ∆H (variación de
la mezcla) es pequeño y ∆F (variación de la energía libre) se hará negativo por –T ∆S
Co-Solventes
(Disolventes)
El cosolvente es el
segundo disolvente añadido al
disolvente original, generalmente en pequeñas concentraciones, para
formar una mezcla que tiene poderes disolventes grandemente mejoradas debido a
sinergismo.
Un solvente que tiene una solvencia
muy alta para un producto, se utiliza en combinación con otro solvente para
obtener un procesamiento más barato. El
efecto resultante es aumentar la solvencia del disolvente bajo precio y
permitir la formulación que contiene un mayor contenido de ingrediente activo.
el cosolvente potenciará el efecto del disolvente activo.
También
se conoce como La mezcla líquida homogénea o de los líquidos en los que el extractante (s) y modificador posible.
Mezclas
Son materiales
que contienen dos o más sustancias simple, que pueden ser separadas tomando
como base las propiedades características de cada una de ellas. Su composición
es variable.
La materia puede
presentarse en dos formas distintas, homogéneas y heterogéneas, según que sean
completamente uniformes, esto es, que sus propiedades y composición sean las
mismas en cualquier punto de la misma o bien que esté formada por dos o más
porciones diferentes, separadas por la superficie definidas a través de las
cuales las propiedades cambian bruscamente.
Un material
heterogéneo es una mezcla y cada porción homogénea de la misma constituye,
desde el punto de vista químico, una fase.
Los componentes
individuales en una mezcla heterogénea están físicamente separados y pueden
observarse como tales. Estos componentes se pueden recuperar por procedimientos
físicos, como la filtración, la decantación o la separación magnética.
En una mezcla
homogénea o disolución el aspecto y la composición son uniformes en todas las
partes de la misma. El componente que está en mayor proporción y que
generalmente es líquido se denomina disolvente, y el que está en menor
proporción soluto.
Las disoluciones
pueden ser sólidas y gaseosas, pero la mayoría de ellas son líquidas. Para
separar los componentes de una disolución se utilizan técnicas como la
cromatografía, la destilación o la cristalización fraccionada.
Si una fase
homogénea puede tener una composición variable se denomina disolución.
Las disoluciones
más utilizadas son las acuosas que el disolvente es el agua.
Las
composiciones de cualquier mezcla heterogénea pueden cambiarse en las
extensiones que se quieran. Pero la composición de una mezcla homogénea, esto
es una disolución, solo puede variarse, en general, entre límites definidos.
Cuando una disolución está en equilibrio con el soluto puro en exceso, se dice
que es saturada. Los componentes de una disolución pueden separarse mediante
cambios de estado en sustancias puras.
Aspectos termodinámicos clásicos de solución
Ejemplo: para la solubilidad de una solución
La
elaboración de gráficos ponderados del logaritmo de la solubilidad en función
de la temperatura absoluta recíproca permite hallar el cambio entálpico
aparente de solución (Happsoln) a partir de la ecuación de van't Hoff (Ec. 1):

Donde R es
la constante universal de los gases (8.314 J mol—1 K—1). Sin embargo en
tratamientos más recientes se han introducido correcciones a la expresión
anterior para disminuir la propagación de errores y por lo tanto diferenciar
entre efectos químicos propiamente dichos y aquellos debidos únicamente al
tratamiento estadístico utilizado en la regresión, lo que ha llevado a utilizar
preferentemente la denominada temperatura armónica media (Thm), la cual
generalmente corresponde a la temperatura absoluta promedio obtenida entre la
más alta y la más baja estudiadas (Krug et al., 1976). La expresión
corregida más utilizada en la literatura (Bustamante et al., 1998) es la
siguiente:

Cuando
no se obtienen comportamientos lineales en la regresión, estos resultados
indican que Happsoln cambia con la temperatura en el intervalo
estudiado, lo cual a su vez, en primer lugar lleva a plantear modelos
polinómicos de regresión de orden dos (Ec. 3: modelos parabólicos):
y=a+bx+cx2
(Ec. 3)
En
los cuales, y = ln X2 y x = T—1. Estas
ecuaciones son derivadas y resueltas punto a punto para hallar Happsoln a
cada temperatura mediante la siguiente expresión:

Una
segunda forma de procesar datos de solubilidad en función de la temperatura
cuando estos no son lineales en la relación de van't Hoff es mediante la
siguiente ecuación presentada por Grant et al. (1984), que también es
conocida como Ecuación de Kirchhoff (Van Ness y Abbott, 1982):

A
partir de la cual, la entalpía aparente de solución se calcula utilizando los
coeficientes a y b de la Ec. 5 mediante la expresión:

La
Ec. 6 puede utilizarse para calcular Happsoln a cualquier
temperatura dentro del intervalo estudiado, sin embargo los resultados son más
confiables cuando esta propiedad se calcula a la temperatura armónica.
En
las ecuaciones anteriores se ha utilizado la concentración del soluto en la
solución (X2) en lugar de la actividad termodinámica correspondiente del soluto
(a2) por lo que en tratamientos más elaborados se han utilizado correcciones
para hacer más coincidentes los valores de entalpía de van't Hoff con los obtenidos
por técnicas calorimétricas, uno de estos tratamientos es el planteado en la
siguiente ecuación (Hollenbeck, 1980; Bustamante et al., 1998):
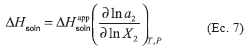
Donde,
el segundo término de la derecha representa la desviación en la entalpía
respecto al valor real debido a posibles variaciones de la actividad
termodinámica del soluto con la concentración del mismo en las soluciones, y se
puede calcular a partir de la Ec. 8 (Manzo y Ahumada, 1990; Bustamante et
al., 1998):

En
la cual, los superíndices "sat" indican la condición de saturación
y 2 es la fracción volumétrica del soluto en la solución la cual
puede hallarse utilizando el concepto de volumen de desplazamiento (D2), el
cual es una adaptación práctica del concepto termodinámico de volumen molar aparente
(Pérez et al., 2003). La expresión más utilizada para
calcular D2 es:

En
la cual, m2 y m1 son las masas (g) del soluto y el
solvente, D1 es el desplazamiento del solvente (densidad recíproca,
cm3 g—1) y r es la densidad de la solución (g cm—3) (Pérez et al.,
2003).
Puesto
que los solutos estudiados en este trabajo son líquidos, lo que conduce a que
la actividad termodinámica de estos en la saturación se considere unitaria
(Yalkowsky, 1999), propiedad que es coincidente con la solubilidad ideal,
entonces se plantea la Ec. 10 para hacer utilizable directamente la Ec. 8
mediante la Ec. 11.


El
cambio de energía libre estándar para el proceso de solución (D Gappsoln) se ha
calculado tradicionalmente como:

El
cual puede también ser normalizado multiplicando por el factor
(ln a2/ln X2) T, P para expresar la función en términos de
actividad termodinámica en lugar de concentración.
En
cambio entrópico estándar aparente de solución ( Sappsoln) se obtiene
directamente a partir de los valores aparentes de entalpía y energía libre
utilizando la expresión:

Los
cuales son corregidos mediante la Ec. 14 para obtener los valores reales:

Solubilidad:
Es una medida de la capacidad de
disolverse una determinada sustancia (soluto) en un
determinado medio (solvente);
implícitamente se corresponde con la máxima cantidad de soluto disuelto en una
dada cantidad de solvente a una temperatura fija y en dicho caso se establece
que la solución está saturada. Su concentración puede expresarse en moles por litro, en gramos por
litro, o también en porcentaje de soluto (m (g)/100 mL).
El
método preferido para hacer que el soluto se disuelva en esta clase de
soluciones es calentar la muestra y enfriar hasta temperatura ambiente
(normalmente 25 C). En algunas condiciones la solubilidad se puede sobrepasar
de ese máximo y pasan a denominarse como soluciones sobresaturadas.
No
todas las sustancias se disuelven en un mismo solvente. Por ejemplo, en el
agua, se disuelve el alcohol y
la sal, en tanto
que el aceite y
la gasolina no se
disuelven. En la solubilidad, el carácter polar o apolar de la
sustancia influye mucho, ya que, debido a este carácter, la sustancia será más
o menos soluble; por ejemplo, los compuestos con más de un grupo funcional
presentan gran polaridad por lo que no son solubles en éter etílico.
Entonces
para que un compuesto sea soluble en éter etílico ha de tener escasa polaridad;
es decir, tal compuesto no ha de tener más de un grupo polar. Los compuestos
con menor solubilidad son los que presentan menor reactividad, como son:
las parafinas,
compuestos aromáticos y los derivados halogenados.
El
término solubilidad se utiliza tanto para designar al fenómeno cualitativo del
proceso de disolución como
para expresar cuantitativamente la concentración de
las soluciones. La solubilidad de una sustancia depende de la naturaleza del
disolvente y del soluto, así como de la temperatura y
la presión del
sistema, es decir, de la tendencia del sistema a alcanzar el valor máximo de entropía. Al
proceso de interacción entre las moléculas del
disolvente y las partículas del soluto para formar agregados se le llama solvatación y si el solvente es agua, hidratación.
Soluciones
Regulares
Es aquella que presenta:
HE ≠ 0 y S E=0
Concepto
empleado para explicar el origen de las desviaciones de la ley de Raoult y su
relación con los coeficientes de la actividad. A partir de la expresión ∆mezcla
G para una solución no ideal.
∆mezcla G= NRT (XALNA+ XBLNB)
Es posible expresar los γ
según
LNA γA= β XB+ LNγB = = β XB2
Ecuaciones De Margales:
aA= γxA=
XAeBx
B2=XAeB(1-x)2ª
PA= (XAeB(1-x)2ª)
P*A
β = 0 ____ grafico PA/
P*A en función de XA
es una línea recta (solución ideal)
β > 0___
mezcla endotérmica, interacciones soluto-solvente desfavorables
β < 0___ mezcla exotérmica, interacciones
soluto-solvente favorables
XA___ la
función PA/ P*A en función de X A tiende
a una línea recta y la función eB(1-x)2ª=
-1
XA
< < 1 la función se aproxima a PA= XAeB
P*A
Con
la forma de la ley de Henry al identificar Kh con eB P*A que es distinta para cada sistema
soluto-solvente
Parámetros
de solubilidad:
Es una medida de la energía
intermolecular. Es la raíz cuadrada de la densidad de energía cohesiva (CED).
CED- Es la variación de energía
molar de vaporización (Ev) por unidad de volumen molar (V1) que mantiene juntas
las moléculas.
CED = 2 = Ev / V1
La determinación de dicho parámetro se suele hacer
de forma experimental:
Medidas directas para líquidos volátiles: a
través de las entalpías de vaporización.
Medidas
indirectas para polímeros: primero asignamos al polímero el
valor del mejor de sus disolventes. Luego se asigna al polímero el
valor del disolvente que más hincha la red polimérica.
Hildebrand propone
el parámetro de solubilidad б, que sería igual a la raíz cuadrada de la energía
necesaria para separar las moléculas de determinado tipo (en cal/mL).
Según esta
teoría mientras más similares son los parámetros de solubilidad de dos sustancias,
mas oportunidad existe que sean miscibles entre si.
Nathan Stolow (estudio
los parámetros de solubilidad) utilizo esta noción en el caso del aceite de lino
antiguo. , estudio el hinchamiento de una capa pictórica al aceite y blanco
plomo (espesor 200 µ-envejecimiento de 27 a32 semanas) en diferentes solventes.
Los solventes que provocan máxima hinchazón
y por ende el parámetro de solubilidad son los mas similares a los de
aceite.
Becerra Mariel
Domínguez Angélica
Domínguez Luisa
García Julio
Patete Elimar
Rivero Yerika
Spósito Odette
Grupo Nº 2
Licenciatura Química
Q01
10° trimestre